pdbeccdutils
This exercise demonstrates the use of the pdbeccdutils - a set of python tools for working with small molecule components in the Protein Data Bank archive.
You can use two main resources as input data for the package. wwPDB CCD contains models of all the ligands e.g. drug molecules, cofactors etc. commonly found in the PDB archive, whereas wwPDB BIRD is composed of biologically interesting polymeric molecules such as peptides or oligosaccharides.
CCD components can be downloaded as a bundle either from wwPDB FTP area, or from PDBe FTP area. The components from the PDBe area are enriched with information about common fragments found in the molecules, their molecular scaffolds as well as links to popular small molecule chemistry databases. You can also use PDBeChem to download individual components from the following link using proper CCD id substitution: ftp://ftp.ebi.ac.uk/pub/databases/msd/pdbechem_v2/H/HEM/HEM.cif.
‘BIRD’ molecules can be downloaded in the *.tar.gz format from the following location: ftp://ftp.wwpdb.org/pub/pdb/data/bird/prd/prdcc-split.tar.gz
pdbeccdutils relies on RDKit for the most of its functionality and as such it is best to be used along with conda environment
Install and activate conda environment with rdkit using the following command:
conda create -c conda-forge -n rdkit-env rdkit python=3.7
conda activate rdkit-env
You can then install the pdbeccdutils either from PYPi or directly from the repository using one of the following commands:
pip install pdbeccdutils
pip install git+https://github.com/PDBeurope/ccdutils.git@master#egg=pdbeccdutils
The pdbeccdutils API contains a number of different modules stored in namespaces with respect to their functionality. All off the modules along with useful tips are documented in the documentation. In this exercise we will go through some of the use cases.
The structure of the package looks roughly like this:
pdbeccdutils
computations
parity_method.py
core
ccd_reader.py
ccd_writer.py
component.py
depictions.py
fragment_library.py
scripts
process_components_cif_cli.py
setup_pubchem_library.py
utils
pubchem_downloader.py
web_services.py
Before we start we need to download a CCD component for demonstaration purposes. We are going to use Heme (CCD ID: HEM).
[1]:
import requests
def download_component(ccd_id):
response = requests.get(f'http://ftp.ebi.ac.uk/pub/databases/msd/pdbechem_v2/{ccd_id[0]}/{ccd_id}/{ccd_id}.cif')
cif_path = f'{ccd_id}.cif'
with open(cif_path, 'wb') as fp:
fp.write(response.content)
return cif_path
hem_path = download_component('HEM')
Structure reading
Structure reading can be done using ccd_reader.py module located in the pdbeccdutils.core module. By default, the molecules comes sanitized using RDKit sanitization procedure and our internal process. However, this option can be turned off by specifying optional parameter sanitize=False to the function.
[2]:
from pdbeccdutils.core import ccd_reader
ccd_reader_result = ccd_reader.read_pdb_cif_file(hem_path)
ccd_reader_result
[2]:
CCDReaderResult(warnings=[], errors=[], component=<pdbeccdutils.core.component.Component object at 0x7f86f7154710>)
CCDReaderResult contains a list of possible warnings and errors that were encountered during the structure parsing. There is also a convenience method that allows reading in multiple chemical components provided they are listed in different data blocks in a single mmCIF file at the same time.
[3]:
components_result = ccd_reader.read_pdb_components_file('HEM.cif', sanitize=False)
components_result
[3]:
{'HEM': CCDReaderResult(warnings=[], errors=[], component=<pdbeccdutils.core.component.Component object at 0x7f86f6804050>)}
Component object
The component object contains a list of usefull properties retrieved directly from the CCD file as well as shorthand functions to the RDKit functionality in the following excercise we will go through some of them.
[4]:
component = ccd_reader_result.component
component
[4]:
<pdbeccdutils.core.component.Component at 0x7f86f7154710>
[5]:
component.formula
[5]:
'C34 H32 Fe N4 O4'
[6]:
component.inchikey
[6]:
'KABFMIBPWCXCRK-RGGAHWMASA-L'
Scaffolds
One of the shorthand function allows you to calculate molecular scaffolds using get_scaffolds() method. You can choose a number of different scaffolds to compute, if you don’t provide any parameter Murcko scaffold is computed by default.
[7]:
# at first scaffolds property is empty
component.scaffolds
[]
# returns an array of rdkit Mol objects with scaffolds found.
component.get_scaffolds()
[7]:
[<rdkit.Chem.rdchem.Mol at 0x7f86f7cb9ad0>]
[8]:
scaffold_details = component.scaffolds[0]
scaffold_details
[8]:
SubstructureMapping(name='MurckoScaffold', smiles='C1=CC2=[N+]3C1=Cc1ccc4n1[Fe-2]31n3c(ccc3=CC3=[N+]1C(=C4)C=C3)=C2', source='RDKit scaffolds', mappings=[['CHA', 'CHB', 'CHC', 'CHD', 'C1A', 'C2A', 'C3A', 'C4A', 'C1B', 'C2B', 'C3B', 'C4B', 'C1C', 'C2C', 'C3C', 'C4C', 'C1D', 'C2D', 'C3D', 'C4D', 'NA', 'NB', 'NC', 'ND', 'FE']])
[9]:
# list of lists of atom names with atoms that are part of the scaffold.
scaffold_details.mappings
[9]:
[['CHA',
'CHB',
'CHC',
'CHD',
'C1A',
'C2A',
'C3A',
'C4A',
'C1B',
'C2B',
'C3B',
'C4B',
'C1C',
'C2C',
'C3C',
'C4C',
'C1D',
'C2D',
'C3D',
'C4D',
'NA',
'NB',
'NC',
'ND',
'FE']]
Fragments
pdbeccdutils package contains also a shorthand functions to enable searching through fragment libraries. By default the code is supplied with a hand-curated library that is internally used by the PDBe as well as collaborating resources ENAMINE and DSI. You can read more about the molecular fragments the built-in library contains in the documentation as well as about an input file format in case you want to supply your own fragment library.
[10]:
from pdbeccdutils.core.fragment_library import FragmentLibrary
# if you dont provide any parameter to the FragmentLibrary constructor
# supplied fragment library is used
library = FragmentLibrary()
fragments = component.library_search(library)
len(fragments)
[10]:
2
[11]:
fragments[1]
[11]:
SubstructureMapping(name='pyrrole', smiles='c1cc[nH]c1', source='PDBe', mappings=((4, 5, 6, 7, 38), (21, 22, 23, 24, 40)))
[12]:
for f in component.fragments:
print(f'{len(f.mappings)} instance(s) of the fragment {f.name} found. SMILES:{f.smiles}')
1 instance(s) of the fragment porphin-like found. SMILES:C1~C~C2~C~C3~C~C~C(~C~C4~C~C~C(~C~C5~C~C~C(~C~C~1~N~2)~N~5)~N~4)~N~3
2 instance(s) of the fragment pyrrole found. SMILES:c1cc[nH]c1
Molecular depictions
component object exposes method compute_2d() that takes DepictionManager object as an argument. This enables you to take advantage of depiction templates supplied with the package. Alternativelly, you can provide paths to your own depiction templates and templates from PubChem. You build a library of pubchem templates using the following script.
[13]:
from rdkit.Chem.Draw import IPythonConsole # this import allows you to display images directly in the jupyter notebook
from IPython.core.display import SVG # allows to display SVG images
[14]:
from pdbeccdutils.core.depictions import DepictionManager
# create an instance of the depiction manager
d = DepictionManager()
result_2d = component.compute_2d(d)
result_2d
[14]:
DepictionResult(source=<DepictionSource.Template: 2>, template_name='hem', mol=<rdkit.Chem.rdchem.RWMol object at 0x7f86f81bd330>, score=0.0)
We can use the score that is part of the result to find out the quality of the depiction. Generally, the lower the better. Higher values indicate bonds crossings and crowded atoms.
[15]:
# depiction score
result_2d.score
[15]:
0.0
[16]:
# and the depiction itself
result_2d.mol
[16]:
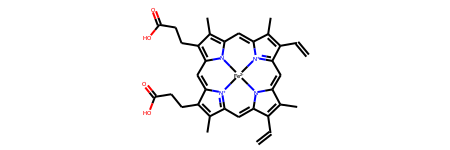
Let’s have a look at the example where molecular depiction cannot be drawn as collision free. We are going to use Adamantone as an example.
[17]:
ado_path = download_component('ADO')
ado = ccd_reader.read_pdb_cif_file(ado_path).component
ado_depiction_result = ado.compute_2d(d)
print(f'Depiction score is: {ado_depiction_result.score}')
ado_depiction_result.mol
Depiction score is: 1.0
[17]:
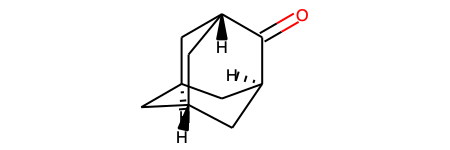
[18]:
# we can also use convenient functions to store the depiction as an svg image:
component.export_2d_svg('HEM.svg')
# including atom names
component.export_2d_svg('HEM_with_names.svg', names=True)
SVG('HEM_with_names.svg')
[18]:

We can also export the svg image and highlight atoms and bonds. Let’s highlight the molecular scaffold we calculated earlier.
[19]:
# retrieve list of atom names that are part of the scaffold
# and color them in green. RGB: (0.2, 0.5, 0.2).
# Scale 0-1 is used instead of the common 0-255
scaffold = component.scaffolds[0]
atom_names = scaffold.mappings[0]
atom_color_mapping = {x: (0.2, 0.5, 0.2) for x in atom_names}
# find out all the bonds that are formed among the scaffold atoms and color them in green as well.
bond_color_highlight = {}
bonds = component.mol_no_h.GetBonds()
for bond in bonds:
begin = bond.GetBeginAtom().GetProp('name')
end = bond.GetEndAtom().GetProp('name')
if begin in atom_names and end in atom_names:
bond_color_highlight[(begin, end)] = ((0.2, 0.5, 0.2))
# draw the final image
component.export_2d_svg('HEM_with_scaffold.svg',
atom_highlight=atom_color_mapping,
bond_highlight=bond_color_highlight)
SVG('HEM_with_scaffold.svg')
[19]:

The exactly same operations can be done by using input data from wwPDB BIRD dictionary. For the following example we are going to use Vancomycin, an antibiotic used for bacterial infection treatment. Just download PRDCC_000204 wwPDB BIRD entry from the FTP directory.
[20]:
c = ccd_reader.read_pdb_cif_file('PRDCC_000204.cif').component
c.compute_2d(d).mol
[20]:
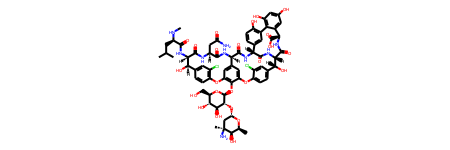
Structure writing
Structure writing can be done using ccd_writer.py module located in the pdbeccdutils.core module. This module exposes write_molecule() method that enables writing out conformers of chemical components in a number of different formats. See example below:
[21]:
from pdbeccdutils.core import ccd_writer
from pdbeccdutils.core.models import ConformerType
# write out ideal conformer in the PDB format
ccd_writer.write_molecule('HEM_ideal.pdb', component, conf_type=ConformerType.Ideal)
# write out model conformer in the SDF format without hydrogens
ccd_writer.write_molecule('HEM_model_no_h.sdf',
component, remove_hs=True, conf_type=ConformerType.Model)
# write out metadata information in CML format
ccd_writer.write_molecule('HEM_in_cif.cml', component)
Parity method
There is a number of chemical similarity methods that can estimate a degree of similarity between the two molecules. pdbeccdutils implements one of such methods that takes advantage of maximum common sub-structures. This method is called PARITY and was developed by Jon Tyzack while working at EBI. You can read the description of the method in the published paper.
In the following example we are going to examine molecular similarity of two heme variants HEME A and HEME D
[22]:
# download the structures first
hem_a_path = download_component('HEA')
hem_d_path = download_component('DHE')
[23]:
from pdbeccdutils.computations.parity_method import compare_molecules
hem_a = ccd_reader.read_pdb_cif_file(hem_a_path).component
hem_d = ccd_reader.read_pdb_cif_file(hem_d_path).component
result = compare_molecules(hem_a.mol_no_h, hem_d.mol_no_h)
result
[23]:
ParityResult(mapping={0: 0, 5: 5, 6: 6, 1: 1, 35: 42, 31: 38, 32: 39, 4: 4, 27: 37, 23: 27, 24: 28, 3: 3, 21: 26, 17: 16, 18: 17, 2: 2, 9: 9, 8: 8, 7: 7, 12: 11, 13: 12, 14: 13, 15: 14, 16: 15, 10: 10, 19: 18, 20: 20, 42: 22, 44: 23, 45: 25, 22: 19, 25: 29, 26: 31, 29: 33, 30: 34, 28: 30, 33: 40, 34: 41, 37: 44, 38: 45, 39: 46, 40: 47, 41: 48, 36: 43}, similarity_score=0.6029411764705882)
ParityResult.mapping contains left to right atom equivalency.
[24]:
print(f'Similarity between heme A and heme D is: {result.similarity_score:.3f}.')
Similarity between heme A and heme D is: 0.603.
Scripts
pdbeccdutils package implements also scripts that we internally use for chemistry related processes in the PDBe. Directly from the package you can try out the PDBeChem that generates content of the FTP area and provides all the data that are consumed by the service.